ENGINEERING the FOLDING of GFP
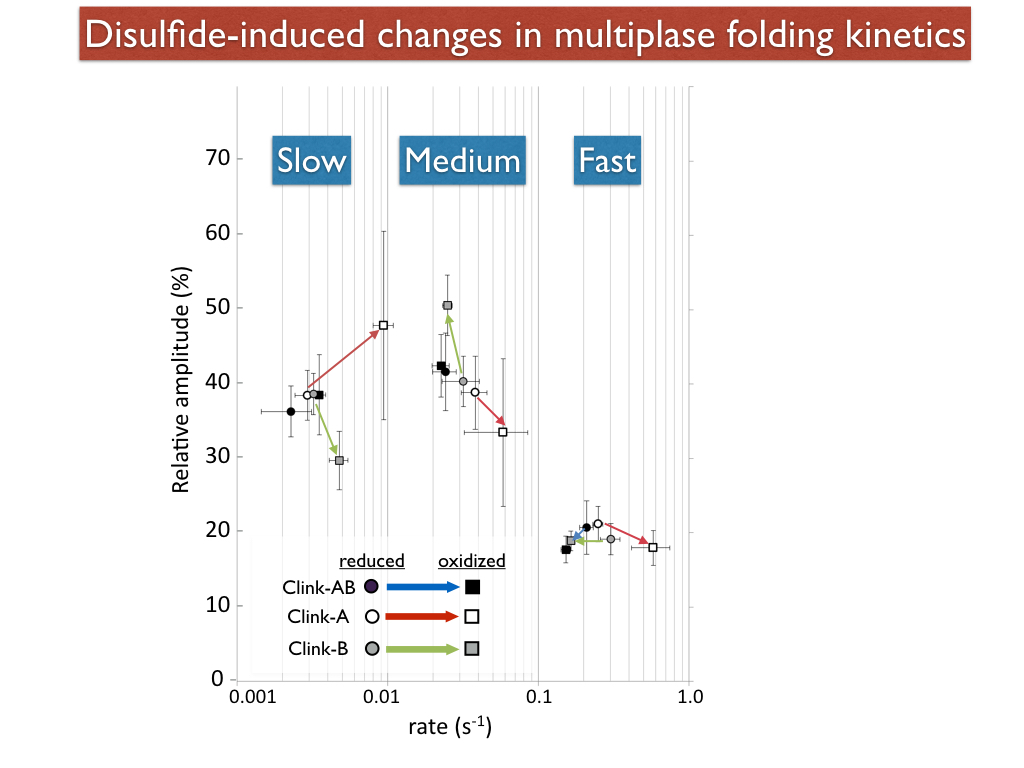
Green Fluorescent Protein (GFP) folds very slowly and show three distinct phases of folding. In a closed barrel protein, the closing of the barrel is probably the last step in the folding pathway. We engineered GFP such that a disulfide bridged two of the gaps (called "seams" in my lab) between beta strands in the barrel. The oxidized versions of these constructs showed differences in kinetics that suggested that the "9-4 seam" (gap between strands 9 and 4) folds last in the fast phase pathway, and that the 7-8 seam folds last in the medium phase pathway. (Pitman et al, 2015)
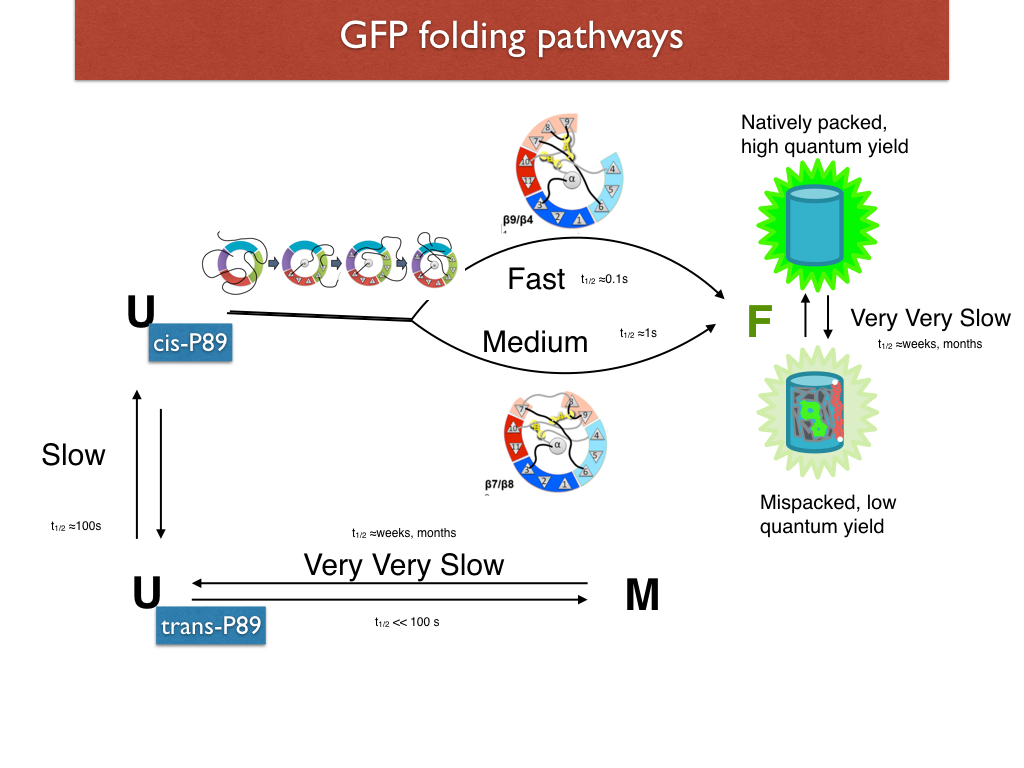
In a separate experiment, we mutated out a loop containing a cis X-Pro and showed the loss of the slow phase of folding. In yet another series of experiments, we systematically omitted each of the strands and showed that the LOO-GFPs varied in their solubility. We attribute the variable solubility to different degrees of nativeness of structure. Combining the evidence from these experiments we have come up with a big picture for the GFP folding pathway. (Rosenman et al, 2014)
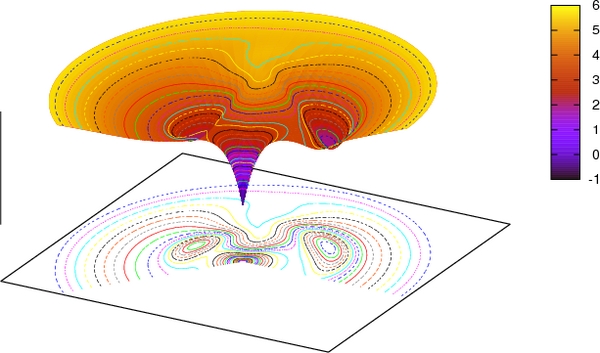
Viewed as an energy landscape, two shallow energy wells exist, separated by a high barrier, and separated by low barriers to the native conformation. Not shown is a very high barrier between natively pack and mispacked closed-barrel near-native conformations, as found by exhaustive mutations in the core. (Banerjee et al, 2017)
CALF
Folding simulations for two small proteins 1LEA and 1RPO using CALF, a knowledge-based force field for C-alpha-only chains. CALF uses probability fields attached to each residue to model backbone-backbone hydrogen bonding. Both simulations show a single replica in temperature exchange Langevin dynamics. (Buck & Bystroff, 2008, 2011)
HMMSTR
By applying data mining techniques and machine learning, we have identified correlations between sequence patterns and local 3D structural patterns, called I-sites. These correlations can be used to predict the protein's three-dimensional structure, either in short fragments or globally using simulations. Molecular dynamics has been used to explain the stability of some short sequences.

We have developed a hidden Markov model called HMMSTR to describe the grammatical structure of conserved sequence patterns within proteins in general. The models can be used to predict protein three-dimensional local structure, secondary structure, to identify protein-coding ORFs, or to design a sequence to fit a structure. The two bioinformatic models described above constitute the first two steps (initiation and propagation) in a five-part heirarchical statistical model for protein folding pathways. The remaining steps (condensation, moltenglobule and sidechain packing) are the subjects of ongoing efforts.
To model the third step, condensation, we have developed a knowledge-based potential that predicts interresidue contacts in proteins. This research direction led to studies into the feasibility of protein structure prediction in two dimensions, the contact map appraoch. Investigations into knowledge-based potentials are continuing by establishing the first sequence-dependent backbone angle and hydrogen-bonding potentials for use in simplified molecular dynamics simulations.
To model the fourth step, we have developed a state-of-the-art method for non-sequential structural alignment of proteins, leading to a database of protein "cores". Future developments of this SCALI database will be to model each core as a hidden Markov model (HMM), for prediction of protein core structures, addressing the "molten globule" state of folding.
To model the final stages of folding, we have developed a mechanistic model called GeoFold.
Contact map predictions can be viewed by browsing the (under construction) HMMSTR-CM database. Contact map predictions can be done for any protein sequence using the HMMSTR server.